Water and Biomolecules
Contents
1 The N-methylacetamide (NMA) dimer
As one can see from the structures and from the following schemes, the N-methylacetamide (NMA) molecule can be understood as one peptide residue terminated with two CH3 ligands. Generally, a peptide chain consisting of amino acid residues looks like this
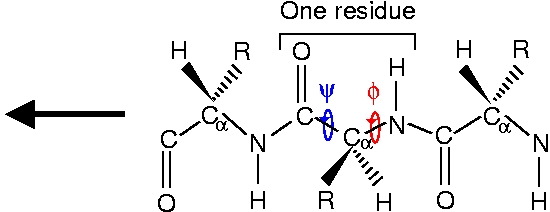
The side chain marked R distinguishes the 20 different amino acids. In general, there are hydrophobic, polar and charged amino acids, depending on the properties of the side chain. The amino acids used in this exercise are glycine (Gly) with R=H and alanine (Ala) with R=CH3.
A dimer consisting of two NMA molecules looks like this
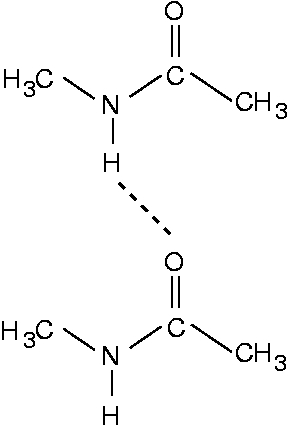
Therefore the NMA molecule corresponds to a Gly residue terminated by two CH3 groups.
The NMA model is a good model for a peptide chain or a β-sheet in the sense that it models the peptide unit well. In the NMA dimer, the most important hydrogen bond, the N-H...O=C bond, is also well modeled. This hydrogen bond is also very important for the stability of β-sheets. On the other hand, also a C-H...O=C hydrogen bond plays an important role in some β-sheets, and this hydrogen bond is not present in an NMA dimer. Another difference is that in the NMA dimer, the two molecules are rather flexible and can orient themselves ideally for forming an N-H...O=C hydrogen bond. In a real peptide chain, however, the residues experience numerous other constraints and can therefore not form ideal hydrogen bonds.
The interaction energy between the two NMA molecules is calculated as
ΔE=ENMA-dimer-2*ENMA-single
where ENMA-dimer is the total energy of the NMA dimer and ENMA-single is the total energy of a single NMA molecule. We can calculate the interaction energy for the different XC functionals, where only PW91 is calculated self-consistently and therefore marked with (sc).
| XC functional | ΔE [eV] |
|---|---|
| PW91 (sc) | -0.32 |
| PBE | -0.30 |
| RPBE | -0.23 |
The negative sign of ΔE means that the two NMA molecules bind to each other. The hydrogen bond mainly responsible for the attractive interaction is the N-H...O=C hydrogen bond. One would expect the interactions of a real β-sheet to be weaker, as the peptide chains are more constrained and therefore cannot orient themselves ideally for the formation of hydrogen bonds. In this sense the NMA binding energy is an upper bound for the binding energies in a β-sheet. Concerning the different XC-functionals one observed that PW91 and PBE give similar results. This is to be expected, as the PBE functional was constructed to be similar to PW91. The RPBE functional gives a significantly smaller binding energy. While the RPBE functional performs very well for surfaces, for hydrogen bonds the PW91 functional is the better one.
Now we plot the difference in electron density of two interacting NMA molecules and of two single NMA molecules with their positions frozen to the ones from the NMA dimer. We use the script diff_density_plot.py and the plot looks like
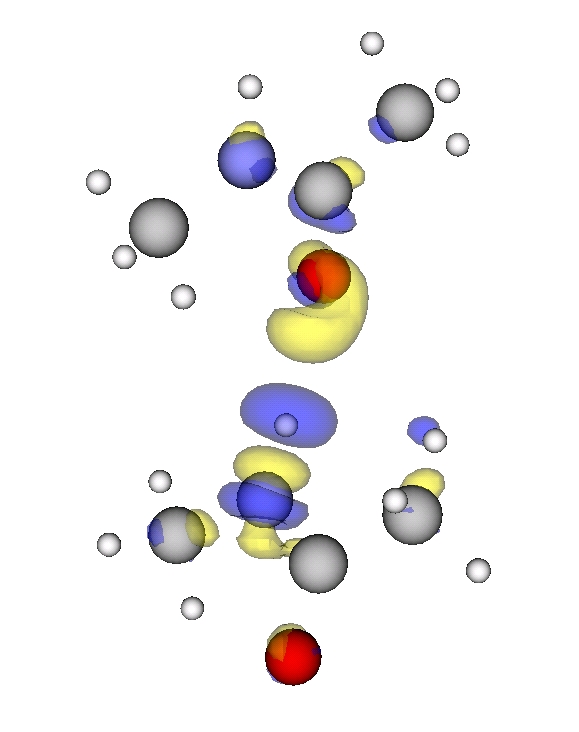
Blue marks the contour for -15 (see note for units) and denotes electron density depletion. Yellow marks the contour at +15 (see not for units) and denotes electron density addition. Thus we can see a clear polarization along the N-H...O=C bond, which confirms that this hydrogen bond is the main reason for the interaction.
2 Parallel and antiparallel two-stranded beta-sheets
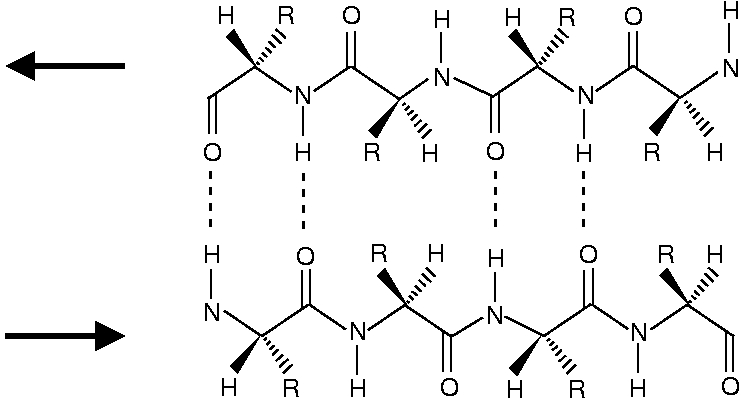
The structure of β-sheets is depited below. The antiparallel β-sheet looks like
and the parallel β-sheet looks like
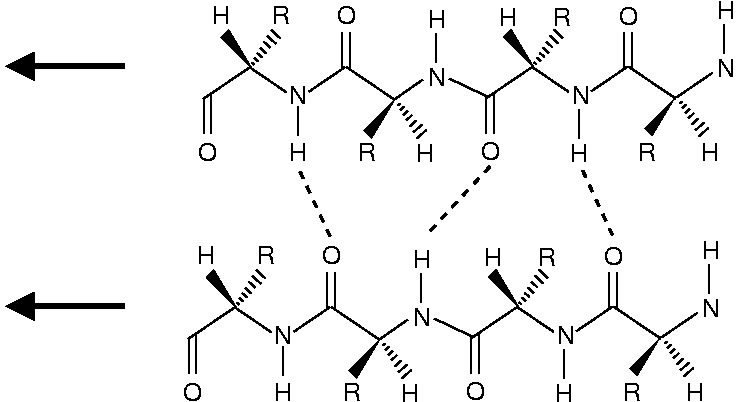
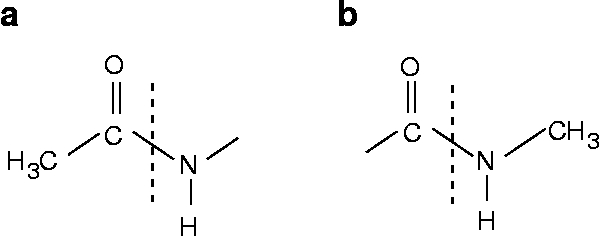
One can see that the peptide chains have been terminated with structure a for the N-end and structure b for the C-end.
One can see that in the antiparallel β-sheet, the peptide chains are ideally positioned for forming a N-H...O=C hydrogen bond. In the parallel chains the N-H...O=C bond geometry is less optimal. As the NMA molecules can assume ideal positions for a N-H...O=C hydrogen bond, the NMA dimer resembles most an antiparallel β-sheet.
The C-N, C-C and C=O bonds in the peptide unit are almost equal for the different β-sheet structures. Therefore, we measure the bond legths for the alanine chains only.
| Bond | antiparallel Ala sheet [Å] | parallel Ala sheet [Å] | organic compounds [Å] |
|---|---|---|---|
| Cα-N | 1.46 | 1.46 | 1.47 |
| N-C | 1.34 | 1.34 | 1.47 |
| C-Cα | 1.54 | 1.54 | 1.54 |
| C=O | 1.23 | 1.23 | 1.20 |
The Cα-N and C-Cα bonds have clearly single bond character. Therefore the peptide chain can rotate around these angles and assume different conformations. The angles describing rotation around these bonds are called dihedral angles. The N-C bond in a peptide chain is significantly shorter than a corr esponding single bond in organic compounds. This shows that the N-C bond in an amino acid has partly double bond charact er and therefore is rigid. In turn, the C=O bond in an amino acid has double bond character, but is slightly longer t han in other organic compounds. Therefore the N-C and C=O bands form two resonating structure with delocalized π-o rbitals. Thus, no rotations around the N-C bond is possible and one peptide unit itself is rather stiff.
For the bonds and angles of the N-H...O=C hydrogen bonds, we concentrate on the Ala peptide chains. We get the following results:
| Parameter | antiparallel Ala sheet | parallel Ala sheet | NMA dimer |
|---|---|---|---|
| NH...OC [Å] | 1.96 | 2.19 | 2.07 |
| N...O [Å] | 2.94 | 3.00 | 3.09 |
| <NH...O [°] | 163 | 147 | 176 |
| <CO...H [°] | 158 | 136 | 144 |
One can see that the bond geometries for the NMA dimer and for the β-sheet structures are nonetheless quite different. This shows that the NMA dimer is useful as a minimum model but limited for bond geometry predictions.
Usually, β-sheet structures are found in hydrophobic regions of the protein. Therefore, hydrophobic amino acids have a bigger probability of being located in a β-sheet than polar amino acids. Therefore, in some situations, water can stabilize β-sheets even more by keeping them together from the outside. It is observed that parallel β-sheets always are buried in hydrophobic regions, whereas antiparallel β-sheets can be exposed to solvent. This suggests that antiparallel β-sheets are intrinsically more stable than parallel β-sheets.
3 The water dimer
The bond length of the water hydrogen bond (O...H) is 1.97 Å. This hydrogen bond is certainly the most important one in nature, as it determines the properties of water.
The two frozen water molecules are calculated with the scripts H2O_freeze1.py and H2O_freeze2.py. The density difference plot is done with the script H2O_density_plot.py and looks like
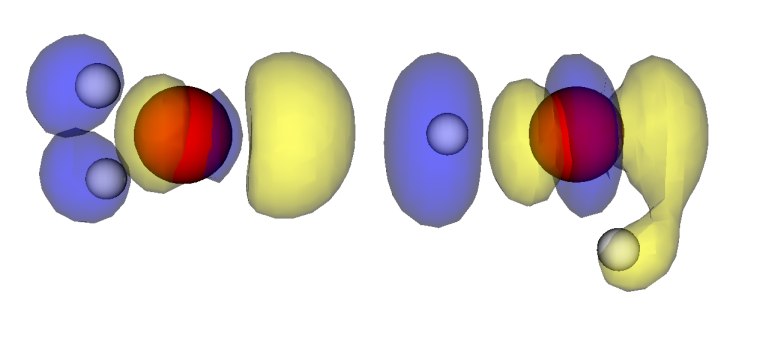
Again, blue denotes electron density depletion and yellow denotes electron density addition.
To calculate the binding energy of the water dimer one has to relax a single water molecule. This can be done using the script 2H2O.py and deleting one water molecule in it. The binding energy is then calculated analogous to the NMA dimer and the result is ΔE=0.23eV for the PW91 functional. This shows that the water molecule binds more weakly than the NMA dimer, but that the binding energies are of the same order of magnitude.