Solution to exercise 2
Contents
1 Aluminum surface and adsorbates
First, we have to find the lattice vectors and stacking sequence of an fcc (111) surface. Two vectors that span the xy-plane of an fcc surface can be found using this drawing:
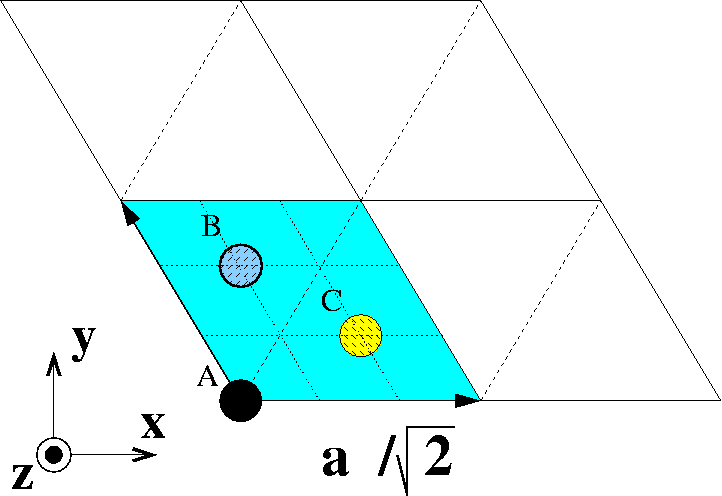
Primitive fcc(111) surface unit cell. Atoms indicate ABCABC... stacking.
The two lattice vectors are:
e1 = (1/21/2, 0, 0)
e2 = (-1/81/2, (3/8)1/2, 0)
The distance between two fcc planes is 1/3 of the body diagonal, therefore it is a/31/2.
The coordination Z of a bulk atom is Z = 12 with six nearest neighbors in the plane, three in the plane above and three in the plane below. As the plane above is removed, a surface atom has a coordination of Z = 9.
When adding the function FCC111 to the script Build.py, we have to think about the stacking sequence. We need a smart way to make sure that the first atom sits at coordinates (0, 0) (in terms of e1 and e2), the second atom at (2/3, 1/3), the third atom at (1/3, 2/3), the fourth atom at (0, 0) and so on. This can be done with the modulo operation, e.g. if n is the number of the layer (starting from zero), the position of the atom is
[(2n % 3)/3, (n % 3)/3, -nd/h]
where d = a/31/2 is the spacing of the layers and h is the total height of the unit cell in the z-direction.
A comment about whether positions should be given in scaled (in terms of the unit cell vectors) or cartesian coordinates: If the coordinates are given before the unit cell is set in the ListOfAtoms, they have to be given in scaled coordinates. If the coordinates of atoms are given at a time, when the unit cell has been set, they have to be given in cartesian coordinates.
1.1 Aluminium fcc(111) Surface Slabs
One can estimate the surface tension from a simple effective medium theory description
Aσ ≃ [1 - (Z/Z0)1/2] Ecoh
The coordination numbers are Z = 9 for the surface and Z0 = 12 for the bulk. The cohesive energy is Ecoh = Eatom - EB = 3.34 eV. The surface area per surface atom is calculated as the shaded area in the figure above. It is
A = a231/2/4 = 7.1 Å2
With these values, one obtains σ = 63 meV/Å2.
The definition of the surface tension is
EN = 2Aσ + NEB
where EN is the total energy of a slab with N layers, A the area of the surface unit cell (the factor 2 because the slab has two surfaces). The limit N -> ∞ corresponds to the macroscopic crystal termination.
Instead of calculating the bulk energy, one can calculate the surface tension from energy differences of different slabs. This has the advantage that there will be a certain error cancellation, as we use the same parameters. One can calculate EN-1 by substituting N by N - 1 in the equation above. By calculating NEN-1 and (N - 1)EN and subtracting them one can show
σ = (NEN-1 - (N - 1)EN) / (2A)
Using a k-point setup of 6 x 6 x 1 one obtains the surface energies
| N | σ [meV/Å2] |
|---|---|
| 3 | 54.9 |
| 4 | 43.2 |
| 5 | 55.8 |
| 6 | 57.5 |
One can see that there is an anomaly for N = 4 but that the surface energy converges for an increasing number of layers. The EMT estimate turns out to be satisfied quite well.
1.2 Adsorption of H on the Al(111) surface
On an fcc (111) surface, there are four inequivalent adsorption sites, as shown:
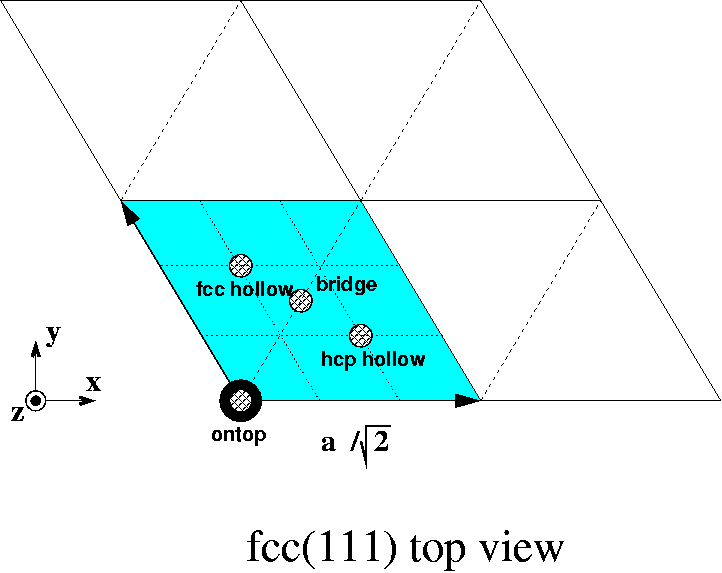
The four different adsorption sites on an fcc(111) surface.
The two hollow sites differ in this way that in the layer below the hcp hollow site there is an atom directly below the hollow site, whereas for the fcc hollow site there is first an atom directly below the hollow site two layers further down.
In scaled (in terms of the unit vectors) coordinates, the four different adsorption sites and their coordination are:
| site | coordinates | coordination |
|---|---|---|
| ontop | (0, 0) | 1 |
| bridge | (1/2, 1/2) | 2 |
| hcp hollow | (2/3, 1/3) | 3 |
| fcc hollow | (1/3, 2/3) | 3 |
In the program, the position of the adsorbate has to be given in cartesian coordinates. The x- and y-coordinates can be calculated using the scaled positions and the unit vectors. For the z-coordinate one has to find a good guess, e.g. from the covalent radii rAl = 1.18 Å and rH = 0.37 Å. One possibility is to assume the Al-H bonds to be the sum of the covalent radii dAlH = rAl + rH. Then the z-coordinate of the adsorbate can be calculated in the following way
| site | z |
|---|---|
| ontop | 1.55 Å |
| brigde | (dAlH2 - a2/8)1/2 = 0.59 Å |
| hollow | (dAlH2 - a2/6)1/2 |
Plugging the values in for the hollow site, one notices that one obtains an imaginary number, i.e. even in the plane the H atom is already further away from the Al atoms than for the covalent radii. However, doing the simulations one notices that the H atom actually ends up sitting ~ 1 Å above the surface. This shows that the rule of thumb with the covalent radii is not always useful. A better approximation for rAl is to use half the distance between the aluminium atoms in the bulk, which is a/2/21/2 = 1.43 Å. Adding this to rH gives dAlH = 1.80 Å. With this it is
| site | z |
|---|---|
| ontop | 1.80 Å |
| brigde | (dAlH2 - a2/8)1/2 = 1.09 Å |
| hollow | (dAlH2 - a2/6)1/2 = 0.71 Å |
which turns out to be better guesses.
Running these calculations with a k-point sampling of 8 x 8 x 1 (probably one can do with less) one gets the relative energies listed here:
| site | total energy [eV] | relative energy [eV] |
|---|---|---|
| ontop | -127.93 | 0 |
| bridge | -128.05 | -0.12 |
| hcp hollow | -128.02 | -0.09 |
| fcc hollow | -128.11 | -0.17 |
The calculation was done with 340 eV plane wave cutoff, 400 eV density cutoff. One can see that the fcc hollow site is the most stable site, followed by the bridge site. In general, the energy differences between the different sites are small. Let us look at diffusion of H from one fcc site to a neighboring fcc site. The H atom will have to pass through a hcp site (transition state), which is 0.08 eV higher in energy. With this activation energy, we can use the Arrhenius equation to find rates:
k = A exp[-Ea/(kBT)]
with A as the prefactor and Ea as the activation energy. The Boltzmann constant is kB = 1.381 x 10-23 J/K. For the different temperatures, we obtain the following rates:
T = 100 K: k = 9.3 x 108/sec
T = 200 K: k = 9.7 x 1010/sec
T = 300 K: k = 4.5 x 1011/sec
T = 500 K: k = 9.8 x 1011/sec
For the rule of thumb for biological catalytic processes, let us assume room temperature to be T = 293 K. The temperature increase is ΔT = 10 K. We start from the Arrhenius equation and use that the ratio should be 2.
2exp[-Ea/(kBT)] = exp[-Ea/(kB[T + ΔT])]
Ea = kBT log(2)(1 + T/ΔT)
Plugging in the numbers, one obtains
Ea = 0.53 eV
This is a typical activation energy in biological catalytic reactions. One can see that it is significantly larger than the diffusion activation energies we consider here.
For a proper investigation of diffusion barriers, one would take a larger unitcell so that the coverage is not as high as it is in this example. Probably, in this example the adsorption positions of the H atoms are dominated by trying to reduce their repulsive dipole interaction (they form a dipole when adsorbing to the Al(111) surface). One would also have to scan the whole diffusion path. In this example one could do this by keeping the x-y positions of the H rigid and just relaxing the z-component. By changing the x-y components by hand, one can scan several paths. For more complicated problems, one would employ the Nudged Elastic Band method.
1.3 Electrostatic potential
The electrostatic potential for H adsorbed in the ontop site looks as shown in this figure:
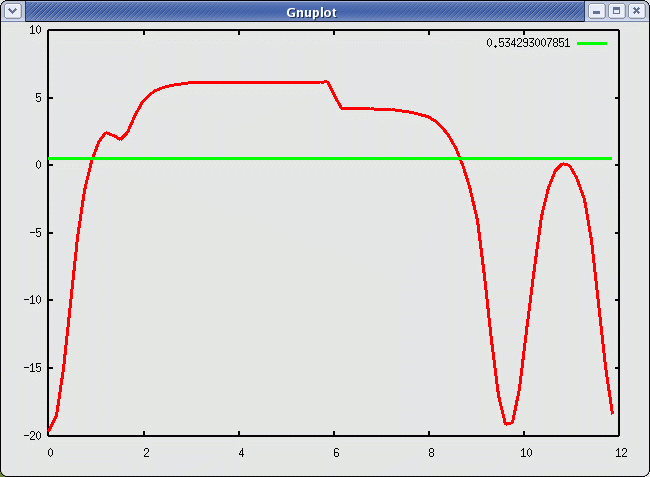
Electrostatic potential and the fermilevel for H/Al(111). This figure has been generated by the script Potential.py.
The H atom is positioned at z = 1.55 Å, and the dipole layer is at z = 6.0 Å. The workfunction for the surface with a monolayer of hydrogen is 5.7 eV and for the clean Al(111) surface we get 3.7 eV. The difference between the two sides of the slab is due to the dipole inducuced by H-adsorption: The H atom pulls electrons away from the surface (μ = -0.078 eÅ). The experimental value for the workfunction of a clean Al surface is 4.25 eV - a bit far from our calculated value of 3.7 eV, but that is to be expected for such a thin slab. For a thicker slab we would get better agreement.
All the number can be found in Dacapos output file:
> grep DIP out.txt DIP: Z_0 [A] 6.000000 nzzero: 41 39 40 42 43 DIP: DIP: u_damp u Field_1 Field_2 Work_f1 Work_f2 V_jump E_dip DIP: [eA] [eA] [V/A] [V/A] [eV] [eV] [V] [eV] DIP: 0.011 0.011 - - - - 0.283 -0.128 DIP: 0.003 -0.142 -0.303 -0.370 - - 0.083 -0.037 DIP: -0.005 -0.180 -0.368 -0.430 - - -0.117 0.053 ... ... ... DIP: -0.077 -0.077 0.012 -0.016 5.663 3.695 -1.965 0.889 DIP: -0.077 -0.077 0.011 -0.016 5.662 3.696 -1.965 0.888 DIP: -0.077 -0.078 0.009 -0.019 5.659 3.693 -1.971 0.891 DIP: -0.077 -0.078 0.010 -0.018 5.663 3.692 -1.974 0.893