Contents
1 Aluminum surfaces and adsorbates
In this exercise, we make a toolbox for building an Al(111) surface. For this surface, we calculate the surface energy and other properties. We will continue to use this surface to investigate adsorbates.
1.1 Making Python Tool boxes
A science project (like the one you are going to make), will often contain some repeated and similar sub tasks like loops over different kind of atoms, structures, parameters etc. As an alternative to a plethora of similar Python scripts, made by copy+paste, it is advantageous to put the repeated code into tool boxes.
Python supports such tool boxes (in Python called modules): put any Python code into a file stuff.py then it may be used as tool box in other scripts, using the Python command: from stuff import thing, where thing can be almost anything. When Python sees this line, it runs the file stuff.py (only the first time) and makes thing available. Lets try an example:
In file stuff.py, put:
constant = 17 def function(x): return x - 5and in file program.py, put:
from stuff import constant, function print 'result =', function(constant)
Now run the script program.py and watch the output.
You can think of ASE and Dacapo as a big collections of modules, that we use in our scripts.
1.1.1 Fcc Surface Builders
As a non-trivial example of a Python module, we'll try to make a tool box for making fcc surface slabs with arbitrary number of layers. A real fcc surface has a large number of atomic layers, but most surface properties are well reproduced by a slab with just 2 - 20 layers, depending on the material and what properties you are looking for.
The most important cubic surfaces are (100), (110), and (111). For face centered cubic, (111) has most compact atomic arrangement, whereas (110) is most open. Here we'll focus on (111), which is most stable.
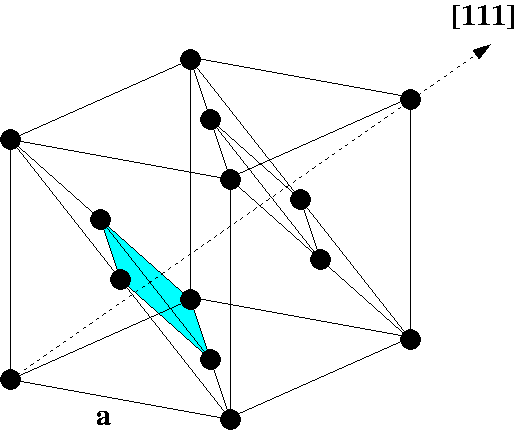
Cubic fcc unit cell
Find two linear independent vectors that span the primitive surface Bravais lattice of fcc(111). Express vectors in Cartesian coordinates x,y,z in terms of the cubic lattice constant a. Please see figure.
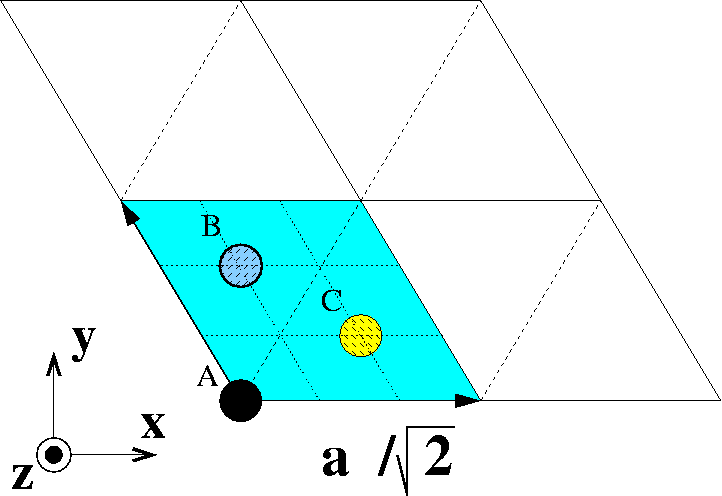
Primitive fcc(111) surface unit cell. Atoms indicate ABCABC... stacking.
What is the coordination Z (number of nearest neighbors) of an fcc(111) surface atom? What is it for a bulk atom?
Now that we know the surface geometry, we can setup a toolbox for making surface structures with arbitrary number of layers. Copy the script Build.py to your area. Browse the script and try to understand it. The central part is the function starting with def BCC100(...) that creates a body centered cubic (100) surface.
Add a function called FCC111 to the Build.py module. This function should build an fcc(111) surface slab. Run the script until you are sure that your new function works (by running the script, you activate the self test in the last few lines of the script, starting at if __name__ == '__main__': - the self test is not done, when you import from your module, though).
hint:
Square roots are calculated like this: 2**0.5 or sqrt(2) (the sqrt function must first be imported: from math import sqrt).
Note
>>> 1 / 3 0 >>> 1 / 3.0 0.33333333333333331
1.2 Aluminum fcc(111) Surface Slabs
In this section, we'll study how surface properties converge, as the slab becomes thicker and thicker.
1.2.1 Surface Energetics
One surface property is the surface tension σ defined implicitly via:
EN = 2Aσ + NEB
where EN is the total energy of a slab with N layers, A the area of the surface unit cell (the factor 2 because the slab has two surfaces), and finally EB is the total energy per bulk atom. The limit N -> ∞ corresponds to the macroscopic crystal termination.
Estimate the surface tension using an expression from the simplest Effective Medium Theory (EMT) description:
Aσ ≃ [1 - (Z/Z0)1/2] Ecoh
where Z and Z0 are the coordination numbers (number of nearest neighbors) of a surface and a bulk atom, respectively, and A is the surface area per surface atom, and Ecoh = Eatom - EB > 0 is the cohesive energy per bulk atom. For Aluminium we have Ecoh = 3.34 eV.
Derive the following equation:
σ = (NEN-1 - (N - 1)EN) / (2A)
Take a look at the script Al111.py. Calculate σ for N = 3, 4 and 5. Use a two-dimensional 6 x 6 Monkhorst-Pack k-point sampling (kpts=(6, 6, 1)). The experimental value of σ is 54 meV/Å2. How well is the EMT estimate satisfied?
1.3 Adsorption of H on the Al(111) surface
One of the most prominent applications of density functional theory is making predictions about alternative atomic structures, when adsorbates are present on the surface. In this exercise, we study the adsorption of H onto Al(111). For simplicity, we use a small and thin Al(111) slab: A 1 x 1 surface unit cell of a two layer Al(111) aurface and deposit a monolayer of H onto the Al(111) slab. The thermodynamics of the H/Al(111) system is governed by the adsorption potential energy surface (PES), i.e. the energy of H/Al(111) as function of the H atom coordinates; in this section, we try to map out and understand the adsorption PES.
- Why are high symmetric sites on the Al(111) surface good candidates for stable adsorption sites?
- How many inequivalent high symmetry adsorption sites are present on a two layer Al(111) 1 x 1 surface unit cell. What is the coordination Z (i.e. number of nearest Aluminum neighbors of an H atom) in each adsorption site?
- There are two inequivalent hollow sites. What is the difference?
- We'll adsorb a monolayer of H in all high symmetry sites of the
Al(111), i.e. one H atom per Al(111) 1 x 1 surface unit cell.
- Use a unit cell with a height of 10.0 Å.
- We relax ionic positions. For simplicity, we'll freeze the Aluminum slab atoms, and only relax the H adsorbate. This is done in the script Relax.py.
- If you want the relaxation to converge quickly, it is necessary to make a qualified guess on the equilibrium position of the H adsorbate. A simple way is to assume that equilibrium bond lengths are the sum of the respective covalent radii. In our case rAl = 1.18 Å and rH = 0.37 Å. You may get these kinds of data at http://www.webelements.com. Select the initial adsorbate elevation over the surface for each adsorption site, so that Al-H bond lengths are 1.55 Å.
Run your script and calculate the total energies of the different adsorption sites, thereby determining their relative energetic stability.
Suppose now that an H atom diffuses from one hollow to a neighboring hollow site at the surface. What is the activation energy for this process? Assuming a prefactor of 1013/sec, how many times does the diffusion take place at T = 100 K, 200 K, 300 K and 500 K.
For biological catalytic processes, a popular rule of thumb is that the rate doubles for every temperature increase of 10 K around room temperature? What activation energy does this correspond to?
If one would want to investigate the diffusion process properly, how would you do this? What would have to be changed from the present setup?
Look at the relaxed configurations with the plottrajectory command:
plottrajectory ontop.nc
Repeat one of your H/Al(111) calculations with slightly more vacuum and use the keyword dipole=True when you create the Dacapo calculator. This will add a dipole layer in the middle of the vacuum region. In order to understand what is going on, take a look at the electrostatic potential and the Fermi-level, when the calculation has finished:
>>> from Dacapo import Dacapo >>> atoms = Dacapo.ReadAtoms('out.nc') >>> calc = atoms.GetCalculator() >>> print calc.GetFermilevel() >>> v = calc.GetElectrostaticPotential() >>> nx, ny, nz = v.shape >>> print nx, ny, nz >>> import Numeric as num >>> vz = num.sum(num.sum(v)) / (nx * ny) >>> import Gnuplot as gp >>> plot = gp.Gnuplot() >>> plot.plot(gp.Data(vz, with='lines'))The workfunction of a surface is defined as the energy required for an electron to escape from the surface. What are the workfunctions of the two surfaces?
1.4 Making nice plots with VMD
One functionality in ASE is that you can make nice plots of the atomic configurations, the Kohn-Sham wave functions and the electron density. Apart from that these plots can be made to look very nice, they can also visualize things which otherwise are hard to analyze or explain. ASE supports visualization tools like VTK, Rasmol and VMD. We will focus on VMD.
1.4.1 Plotting the atoms
Copy the script atomplot.py to your area. Read it and try to understand what it does. Then type python to get a Python prompt and run the script interactively, i.e., cut and paste the commands from the script to the Python prompt. Three windows pop up, an OpenGL display where the atoms are visible, a vmd console, and VMD main. The VMD main window have different menues, open the /Graphics/Representation/ menue and change the drawing method to CPK. VMD can do many things but you should try to use the Render option to make a ray tracing figure of your slab, change the colors of the atoms using different representations, remove the axis indicator and change the background color. Open the cube file containing the density and make a representation that shows a density isosurface. When you have made a povray plot you can use your favorite graphics program (gimp is a good one), to edit your plot and save it as an .eps file, which you can include in latex.
1.4.2 Using VMD to plot density differences
It is sometimes usefull to look at density changes when studying for instance adsorption reactions. Copy the script densitydiff.py to your area. Read it and try to understand what is does. Change the nescesary lines to look at one of your slabs with H adsorbed. There is one major assumption in the script if this is used for the H adsorbed on a metal surface, try to identify it. When you have written the density different to a file open this file in vmd and use it to investigate what is happing.
1.4.3 Using VMD to make input files
VMD is very usefull for setting up input files to your calculations. Use /Mouse/Move/Atom to move H to another position and save the coordinates as .xyz. .xyz files can be transformed into trajectory files with the command xyz2trajectory. Do this and and then use vmd_input.py to start your calculation from the trajectory file. Remember to set your UnitCell and your Calculator