Solution to Exercise 1
Getting started with Dacapo
Contents
1 Bulk Al
1.1 Running a calculation
- The default exchange correlation functional used by Dacapo is the Perdew-Wang 91 GGA functional.
- Taking the weighted average over all k-points one would expect that one band is fully occupied and one band is partly occupied, as Aluminum has three valence electrons. Of course, the occupation numbers vary over the different k-points.
1.2 Convergence in k-points and planewave energy cutoff
For the convergence with respect to k-points, one gets the diagram shown here:
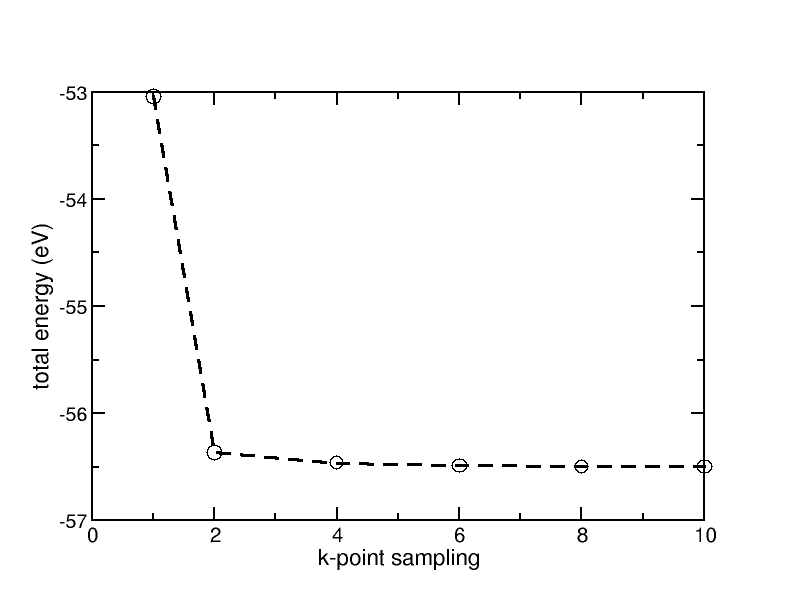
Convergence of the total energy of bulk fcc Aluminum with respect to the k-point sampling
From this one can see that a k-point sampling of 6 x 6 x 6 (6 k-points in each direction of the reciprocal unit cell) should be sufficient. As the last part of the exercise (relative stability of the fcc and the bcc lattice structures) requires a good k-point sampling, we will use a sampling of 10 x 10 x10 in the following.
For the convergence with respect to the kinetic energy cutoff of the plane waves, one gets this diagram:
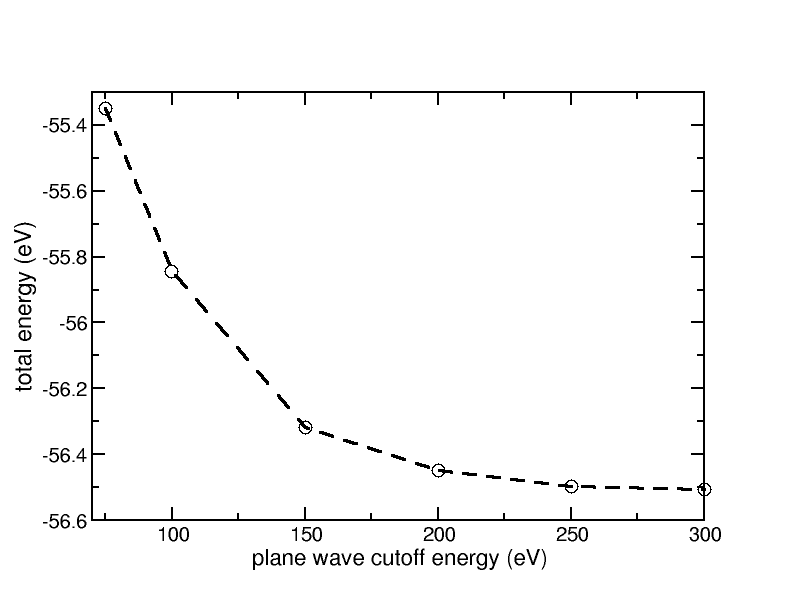
Convergence of the total energy of bulk fcc Aluminum with respect to the kinetic energy cutoff of the plane waves
From this we see that a kinetic energy cutoff of 200-250 eV is sufficient for Aluminum. In the following we use a cutoff of 250 eV.
- The necessary energy cutoff depends on the softness of the pseudopotential and is therefore the same for all Aluminium structures. For other chemical elements, other pseudopotentials with different properties are employed, and therefore, a different energy cutoff may be required. For example, transition metals have often pseudopotentials that require an energy cutoff of 25-30 Ry = 340 - 400 eV. The necessary k-point sampling depends on the complexity of the Fermi surface and the bandstructure. This can be assumed to be similar for different Aluminum bulk structures, and therefore, the same k-point sampling can be used. For other chemical elements, the required k-point sampling is different. In general, metals require a higher k-point sampling than semiconductors or insulators.
- The energy cutoff determines the number of basis functions, which represent the density and the Kohn-Sham wavefunctions. Therefore, the energy cutoff is subject to the variational energy principles, and the total energy of a system has to decrease if the cutoff energy is increased. The necessary k-point sampling depends on the complexity of the Fermi surface. Therefore, one cannot predict, whether the total energy decreases or increases for a finer k-point sampling.
- Al is an easy element to work with, because its pseudopotential is softer than many other metals. It is not magnetic, and therefore spin-polarized calculations are not required. Furthermore, its electronic structure resembles the free electron gas, and therefore convergence is easy and fast.
2 Equilibrium lattice properties
First, we map out the cohesive curve E(a) for Al(fcc), i.e. the total energy as function of lattice constant a, around the experimental equilibrium value of a0 = 4.05 Å. We obtain the curve shown in the following figure, which we fit to a third-order polynomial.
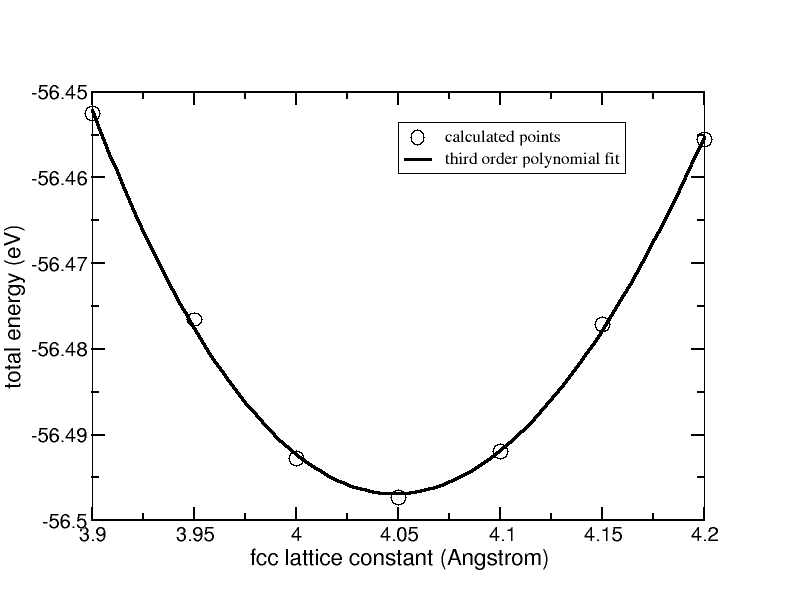
Total energy versus the lattice constant for bulk fcc Aluminum. The solid line is a third-order polynomial fit.
The next step is to take the first and the second derivative of the third order polynomial fit. Now we can easily determine the equilibrium lattice constant a0 by reading off, at which lattice constant the first derivative is zero. Then the value of the second derivative at a0 has to be read off. The results are:
a0 = 4.05 Å and d2E/da2 = 3.84 eV/Å2
For calculating the bulk modulus using the expression
B = M/(9a0)d2E/da2
we note that M, which is the number of atoms per cubic unit cell, is M = 4 for an fcc lattice. Remembering that 1 GPa = 109 J/m3 we have to convert the second derivation from eV/Å2 to J/m2. This results in:
d2E/da2 = 3.84 eV/Å2 = 3.84 x 1.602 x 10-19 J / (10:su p:-10 m)2 = 61.52 J/m2
Keeping in mind that giga corresponds to the prefactor 109, we obtain for the bulk modulus:
B = 4/(9 x 4.05 x 10-10 m) x 61.52 J/m2 = 67.51 GPa
These results compare well to the experimental values a0 = 4.05 Å and B = 76 GPa. The bulk modulus compares not as well as the lattice constant. As it depends on the second derivative of the fit, it is a rather sensitive quantity.
3 Equilibrium lattice properties for bcc
First, we try to find a qualified starting guess for abcc from the value of afcc. One possibility is to assume that the primitive unit cell volumes of the fcc and bcc structure are equal. The unit cell volume for the cubic unit cell of fcc is afcc3 and there are four atoms in the cubic unit cell. Therefore the unit cell volume of the fcc primitive unit cell is afcc3/4. In the same way, one obtains the unit cell volume for the bcc primitive unit cell, which is abcc3/2. Thus
abcc = afcc 2-1/3 = 3.21 Å
Another possibility is to assume that the nearest neighbor distances in the fcc and the bcc structures are equal. The nearest neighbor distance in fcc is half a face diagonal, afcc/21/2. The nearest neighbor distance in bcc is half a body diagonal, (31/2/2)abcc. From this we obtain
abcc = (2/3)1/2afcc = 3.31 Å
As the lattice constant for the bcc lattice is smaller, one should use a higher k-point sampling to keep the product of lattice constant and number of k-points constant. The cohesive energy curve for the bcc Aluminum structure is shown here:
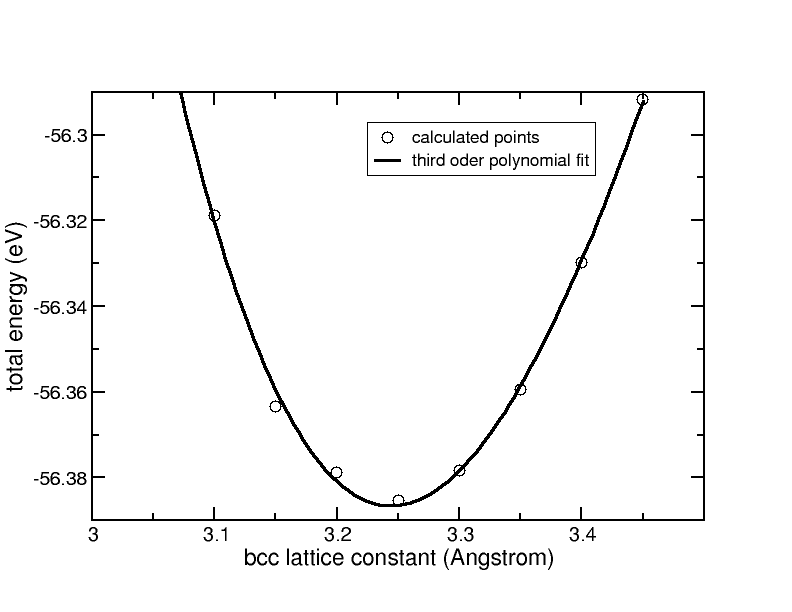
Total energy versus the lattice constant for bulk bcc Aluminum. The solid line is a third-order polynomial fit.
From a third order polynomial fit, we obtain:
a0 = 3.25 Å and d2E/da2 = 5.56 eV/Å2
This gives a bulk modulus of B = 60.9 GPa
The bcc bulk modulus is very similar to the fcc one. One would expect this, as both structures have approximately the same density, and therefore one would expect the elastic properties to be fairly similar.
Now, we calculate the fcc/bcc total energies at their respective lattice constants and for two different plane wave cutoffs: 200 eV and 450 eV. We obtain these values:
| cutoff [eV] | Ebcc [eV] | Efcc [eV] | Ebcc - Efcc |
|---|---|---|---|
| 200 | -56.342 | -56.448 | 0.107 |
| 450 | -56.398 | -56.509 | 0.111 |
One can see that the energy difference between the bcc and the fcc structure is well converged at 200 eV, whereas the total energy is not yet fully converged. In general, energy differences converge much faster than total energies. One can use that fact to reduce the cutoff energy, because convergence of energy differences is all that is needed. The absolute total energy values depend on the pseudopotentials and do not have any physical significance.